
メラノーマに対する分子標的薬として、変異型BRAFを抑制する低分子化合物が開発されており、この BRAF阻害薬は、BRAF変異を持つ転移性メラノーマ症例に対して高い奏効率を示し、全生存期間や無増悪生存期間を有意に延ばしたしかし、半年ほど経つと、BRAF阻 害薬に対する薬剤耐性を示すメラノーマが再び増殖を始めてしまう。このような現象は多くのがん治療においてみられ、がん治療における課題となっている。
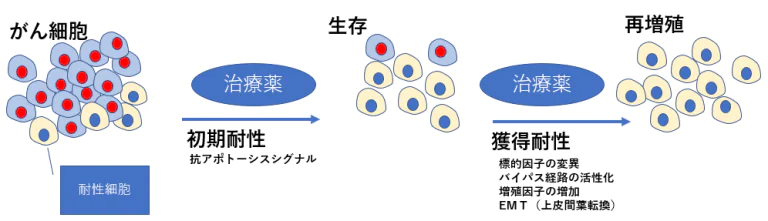
治療初期にがん細胞が死滅しない」という現象に深く関わるのは、抗アポトーシスシグナルである
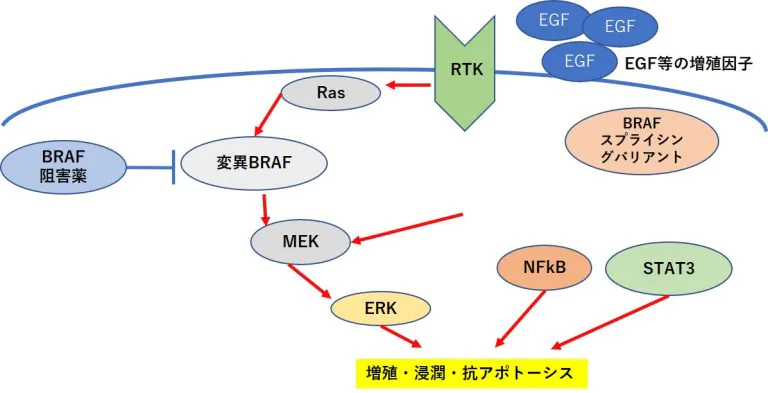
RAFタンパク質→MEK (meiosis-specific serine/threonine-protein kinase)→ERK(extracellular signal-regulated kinase)という一連のキナーゼ タンパク質を順に活性化する経路であるMAPK(mitogenactivated protein kinase)経路が活性化されることで、リン酸化を受けて活性化したERKタンパク質は、抗アポトーシス因子で あるMcl-1(myeloid cell leukemia 1),Bcl-2(B-cell leukemia/lymphoma 2),Bcl-XL(B-cell lymphoma extra-large)の発現を促進する(2、3)。
Mcl-1は、Bclファミリーに属するタンパク質をコードしており、アポトーシスを阻害し細胞生存を促進する。
これらの抗アポトーシス因子はMAPK 経路だけでなく、NFκB(nuclear factor kappa B)やSTAT3 (signal transducer and activator of transcription 3)によっても発現誘導される(4)
EGFR(epidermal growth factor receptor) →RAS(rat sarcoma viral oncogene homolog)→RAF→MEK →ERKの一連の増殖シグナルの異常活性化が主要因であるがん細胞において、この経路を遮断する分子標的薬は STAT3の活性化を引き起こし、抗アポトーシス因子の発現 を促すことで薬剤耐性が生じることが報告されている(5)
PDGFRβ(platelet derived growth factor receptor beta) 量の増加、NRAS(NRAS proto-oncogene, GTPase)変異も BRAF阻害薬耐性と関連している場合がある(6,7)。
- BRAF→MEK→ERK経路を遮断して抗アポトーシス(生存 促進)シグナルを阻害しても、STAT3が活性化することで 再び抗アポトーシス因子の発現が誘導され、アポトーシス が回避されることになる。このようなメカニズムにより、 一部のがんは分子標的薬の存在下でも生き残り、さらに下 記のメカニズムを獲得したがん細胞は再び増殖を開始する。
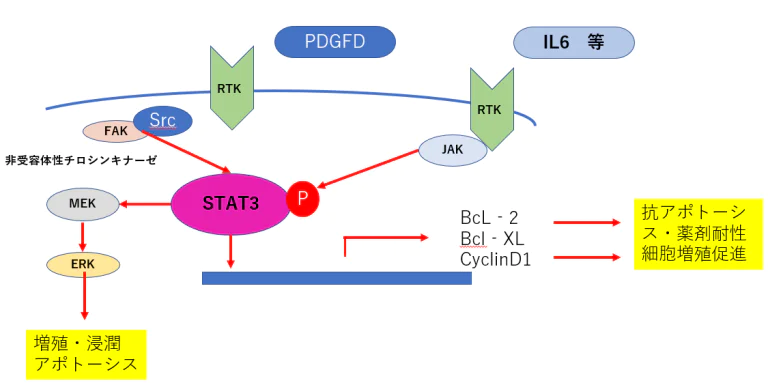
- 肺がん細胞や乳がん細胞においてRTK阻害薬を添加すると、FGF(fibroblast growth factor)やIL6(interleukin 6)の 発現が亢進し、FGFシグナルやIL受容体からのシグナル伝達経路が活性化し、それによるSTAT3の活性化により 抗アポトーシスシグナルが活性化されることが報告されている(5)。
- STAT3により抗アポトーシス経路が活性化し、薬剤耐性を亢進させると考えられる。
増殖シグナルの再活性化に対してP16 の重要性
「薬剤の存在下で再びがん細胞が増殖する現象」には、増殖シグナルの再活性化が関与する。BRAF阻害薬により阻害されるBRAF→MEK→ERK経路は、抗アポトーシ ス因子の発現を誘導するだけでなく、増殖促進シグナルとして働いている。BRAF阻害薬は変異型BRAFに起因するERKの異常活性化を阻害することで効果を発揮するが、BRAF阻害薬の存在下でも、ERKを活性化する機構が生じてしまう。
p16(INK4A) inhibits the pro-metastatic potentials of osteosarcoma cells through targeting the ERK pathway and TGF-β1
Gabriela Silva 1, Abdelilah Aboussekhra 1
・ 2016 May;55(5):525-36. doi: 10.1002/mc.22299. Epub 2015 Mar 1
Gabriela Silva 1, Abdelilah Aboussekhraらの研究によれば
- 細胞外シグナル調節キナーゼ(ERK)は、進化的に保存されたマイトジェン活性化プロテインキナーゼシグナル伝達経路の下流成分であり、様々な生理学的プロセスに関与する多数の遺伝子の発現を制御する。この経路は、最も一般的な原発性悪性骨腫瘍骨肉腫を含む異なるタイプのヒト癌における突然変異または異常な細胞外シグナル伝達によってしばしば過剰活性化される。p16(INK4A)は、骨肉腫において頻繁に失われる重要な腫瘍抑制遺伝子であり、これらの悪性腫瘍の進行と関連しています。
- ここで、ERK1/2プロテインキナーゼが、骨肉腫細胞および正常ヒトおよびマウス細胞においてp16(INK4A)ダウンレギュレーションによっても活性化されることを示した。この阻害効果は、上流キナーゼMEK1/2の抑制と関連しており、miR-21-5pの抑制およびその結果生じる骨肉腫細胞におけるMEK/ERKアンタゴニストSPRY2のアップレギュレーションを介して媒介されていることが示されている。
- さらに、p16(INK4)がERK1/2のmiR-21-5p依存性阻害により、これらの細胞の遊走/浸潤能を阻害することを示した。
- p16(INK4)がTGF-β1の発現/分泌の阻害を通じて間質線維芽細胞に対する骨肉腫細胞のパラクリン遊走促進作用を抑制するという明確な証拠を提示している。
- この効果はERK1/2依存性でもあり、p16(INK4)およびERK1/2は、細胞自律作用に加えて、非細胞自律がん関連機能も有することを示している。
- これらの結果は、腫瘍抑制因子p16(INK4)タンパク質が、細胞周期調節因子としてだけでなく、発癌性/転移促進性経路の負の調節因子としても、骨肉腫細胞の発癌過程を抑制することを示している。これは、ERK経路を標的とすることが最大の治療的価値があることを示している。
上皮間葉転換(EMT)と薬剤耐性の関係
- 膵がんモデルマウスにおいても、ゲムシタビン(抗がん剤の一種)に対する感受性が亢進することが示されている(8)。EMTはがん細胞の薬剤耐性 (抵抗性)を促進する分子機構であることが示されている。
- 乳がんモデルマウスにCTX(cyclophosphamide;抗がん剤の一 種)を投与すると、肺転移巣に存在するがん細胞はほとんどがGFPポジティブな細胞であった。EMTを起こした細胞が抗がん剤存在下でも生き残り、転移を起こしたと考えられる(9)。
上皮間葉転換(EMT)ががんの転移に関与する
- 上皮系の性質を持つ細胞が間葉系の性質を持つ細胞へと変化する現象である上皮間葉転換(epithelial-mesenchymal transition:EMT)は、正常組織発生においてのみならず、がん組織内において もさまざまなシグナルによってEMTが誘導される。EMTを誘導するシグナルとして、TGFβ(transforming growth factor beta)シグナル、Wntシグナル、インテグリンシグナルなどが知られている。がん組織内において、がんの浸潤先端などの一部の部位でEMTは誘導され、がん不均一性の要因となる。
- 2018年にPastushenkoらは、がん転移において上皮と間葉 だけでなく、その中間の性質を持つがん細胞が出現していることを明らかにした10)完全に間葉系の性質を獲得したがん細胞よりも、上皮間葉転換の中間体がより転移を起こすことが示唆されている10)。
- EMTプログラムによりがん細胞の転移だけでなく、幹細胞性も促進される11)さらに、幹細胞性質が亢進すると、薬剤やガンマ線照射などの治療に対しての抵抗性も増す。
上皮-間葉移行(Epithelial-to-Mesenchymal Transition 「EMT」)とphenotype
上皮様(epithelial)から転移可能もしくは準安定(metastable)細胞を経て間葉系(mesenchymal)の細胞へ変化
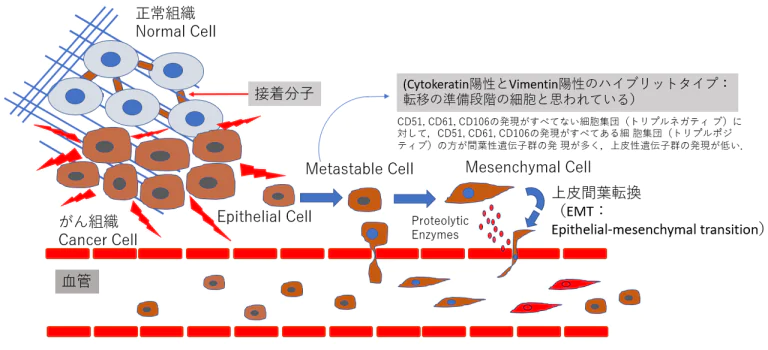
- 乳がんの細胞群をマウスの静脈内に投与し、肺へ の転移能を調べたところ、リプルネガティブ(CD51、CD61、CD106)な細胞群で 転移が顕著に多く観察されている。したがって、完全に間 葉系の性質を獲得したがん細胞よりも、上皮間葉転換の中 間体がより転移を起こすことが示唆されている(9)。
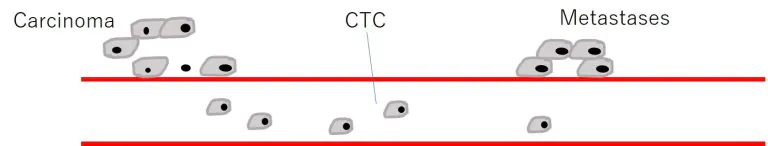
血中循環腫瘍細胞「Circulating Tumor Cells」=CTC
- 血中内を循環するCTCは治療すべき重要なTARGET
その他、薬剤耐性に関与するメカニズム
- 物質の輸送を行うタンパク質である膜貫通型タンパク質トランスポーターの一種であるABCトランスポーターは分子内によく保存されたATP結合領域を持ち、ATPの加水分 解を利用して物質の輸送を行う。薬の排出にも関与していることから、ABCB1はさまざまながんで薬剤耐性に関与していることが報告されている。
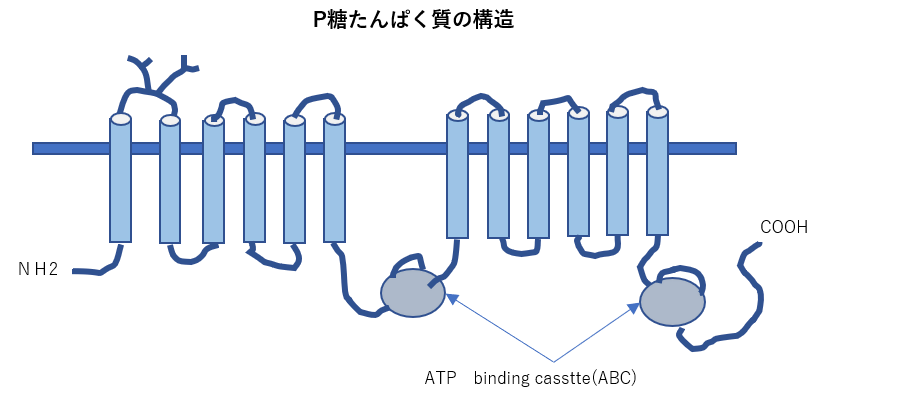 P-glycoprotein(MDR1):
P-glycoprotein(MDR1):植物アルカロイドや抗がん剤を含む多くの疎水性薬剤を輸送します。
Multidrug resistance protein(MRP):
GSH抱合ロイコトリエンC4(LTC4)や17βエストラジオール-17-β-Dグルクロニド、硫酸エストロンのような広い範囲の抱合型親水性分子を輸送します。
薬物濃度の低下=細部内への取り込みの減少と細胞外への排出の亢進
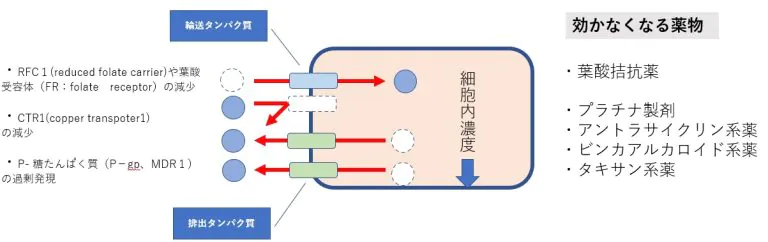
殆どの生物に存在する膜貫通型ATP結合カセット(ABC)輸送体タンパク質ファミリーの増加が挙げられます。これらのタンパク質は、悪性細胞から幅広い様々な治療化合物を積極的に排出することにより、がんの化学治療法耐性を引き起こします。ABC輸送体は様々な細胞や組織、血液組織関門中で毒性化合物に対する重要な保護機能の役割をします。
薬物代謝の変化=活性化の低下と不活化の亢進
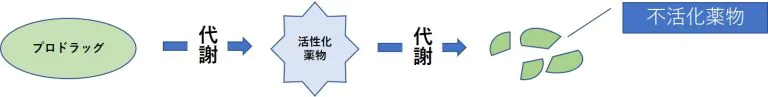
- 体内で代謝を受けて活性化する薬物(プロドラッグ)では、代謝酵素活性が低下すると作用が減弱する。
- 薬物を不活化する酵素の活性が上昇すると作用が減弱する。

※横スクロールで全体を表示
| ・各種代謝酵素の活性低下 |
効かなくなる薬物フッ化ピリミジン系薬 |
| ・DPD(ジヒドロピリミジン脱水素酵素:dihydropyrimidine dehydrogenase)の活性上昇 | フッ化ピリミジン系 |
| ・GST(glutathione S-transferase)の活性上昇 | ナイロジェンマスタード系 (多標的阻害薬) |
| ・シチジンデアミナーゼの活性上昇 | シタラビン |
| ・TPMTの活性上昇(チオプリンメチルトランスフェラーゼ:thiopurine methyltransferase) | 6-MP |
標的分子の量的変化による薬剤耐性①
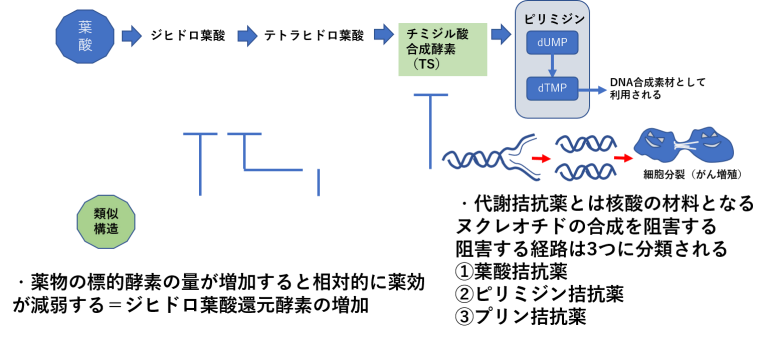
標的分子の量的変化による薬剤耐性②
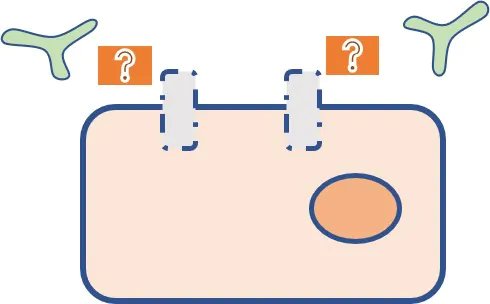
- 分子標的薬では標的抗原の発現がなくなると作用できなくなる。
効かなくなる薬物
- 各種抗体薬
標的分子の質的変化による薬剤耐性
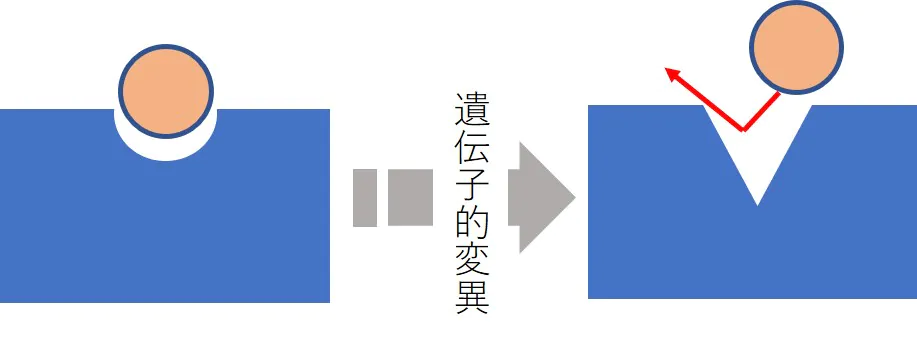
- 分子標的薬では標的タンパク質の遺伝子変異などにより、構造が変化して薬物が結合できなくなる。
効かなくなる薬物
- トポイソメラーゼの変異=トポイソメラーゼ阻害薬
- βチューブリン=タキサン系
- チロシンキナーゼの変異=各種TKI
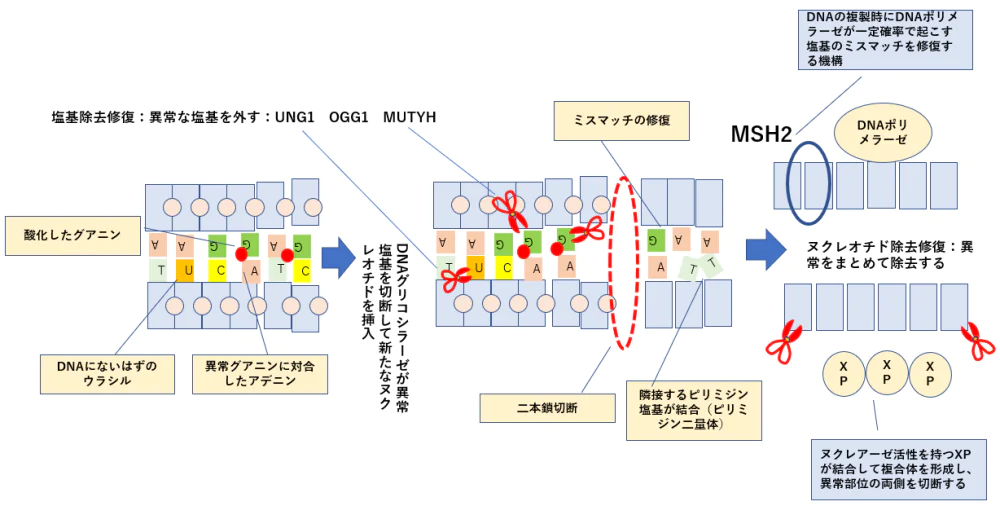
細胞死に対する耐性=DNA修復機構の亢進
- 薬物がDNAを障害しても修復されてしまうために細胞死が起こらなくなる。
ERCC1(DNA架橋除去酵素の発現) プラチナ製剤の抵抗性
UNG1の亢進 フッ化ピリジミン系薬の抵抗性
MGMTアルキル基除去酵素の過剰発現 アルキル化薬の抵抗性
アポトーシス抵抗性
- 多くの薬物はがん細胞にアポトーシスを誘導することで効果を示すため、一方で、化学療法の継続が、アポトーシスの抵抗性を形成してやがて効果が低下する。
P53 の異常(変異、メチル化)が薬剤耐性をもたらす
P53のメチル化などにみられる機能的なp53の喪失は、化学療法薬に対する耐性の増加をもたらす。MDR1を通常発現する腫瘍における多剤耐性遺伝子(MDR1)発現は、変異型p53に依存的に調節されるものである。野生型p53は、内因性mdr1遺伝子発現を調節する。
癌に見られるp53不活性化は、MDR1発現のアップレギュレーションのために化学療法剤に対する選択的耐性をもたらす。
p53の変異は、p53発現腫瘍における化学療法耐性(14-16)および再発(17)と相関し、p53の不在は、エトポシド、アドリアマイシン、および5-フルオロウラシルなどの化学療法剤に対する耐性と関連している(13)
p53の変異は、p53発現腫瘍における化学療法耐性(2-4)および再発(17)と相関する。
P53 を正常に機能させるためにはCdc6たんぱく質をRNA干渉によってその発現を減少させることやメチル転移酵素の分解といった複合的な治療により効果が異なっていることが判明してきました。
(分化療法)を参照
抗アポトーシスBCL2の発現抑制のための遺伝子治療
EZH2RNA干渉治療=EZH2ノックダウン療法
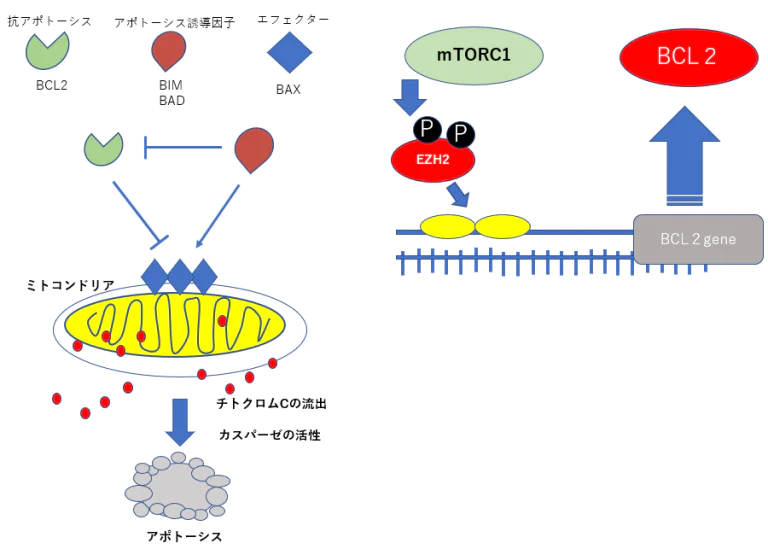
BCL2の発現を抑えるためにEZH2RNA干渉によってEZH2たんぱく質の産生を妨げる。 mTORの発現はEZH2を活性化するのでこの上流のTHKのシグナルの抑制にPTEN、P16を効果的に使用する
- 1)EMT を誘導するシグナルとして,TGFβ(transforming growth factor beta)シグナル,Wntシグナル,インテグリンシグナ ルを抑制する。=KLOTHO/Cdc6shRNA
- 2)増殖シグナルの再活性化に関するERKを抑制=P16
- 3)PIP2~PIP3の脱リン酸化とNFkBを抑制させる=PTEN
KLOTHO-encordingRNA/Cdc6nkshRNA P16/PTEN‐encordRNA=を含有したecmRV-RNA治療
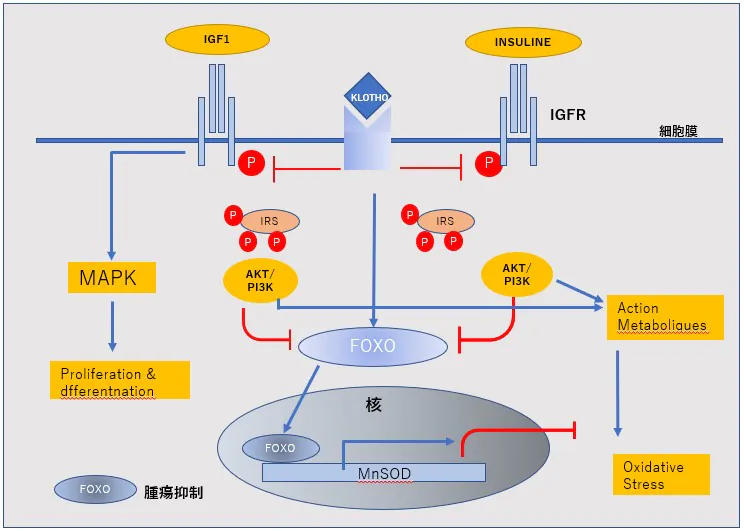
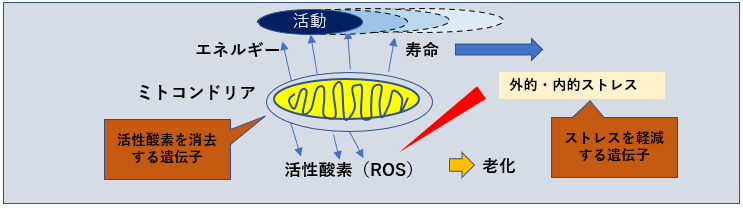
SODは、スーパーオキシドアニオン(O2)を酸素と過酸化水素(H2O2)へ変換する酸化還元酵素。
銅イオンと亜鉛イオン(Cu, ZnSOD)、マンガンイオン(MnSOD)、鉄イオン(FeSOD)などの金属イオンを持った酵素。
細胞質(Cu, ZnSOD) やミトコンドリア(MnSOD)に多く局在している。
寿命と相関するSOD活性
酸素消費量の多さに対してSOD活性の強さは、寿命と相関があると言われている。 SOD1は、細胞液中 SOD3は細胞外に存在する。
SOD-2(MnSOD)
ミトコンドリア内に存在し、ミトコンドリアで産生されたスーパーオキシド(ーO2)を毒性の低い過酸化水素と酸素に変換。
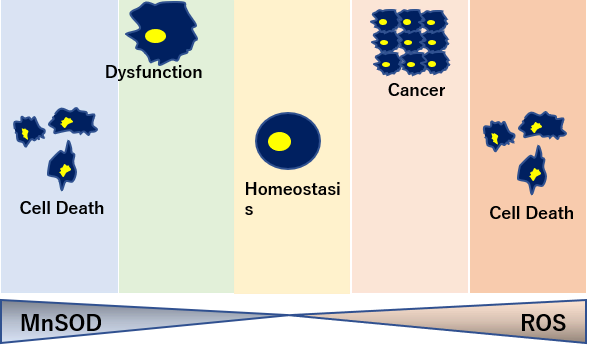
トレードオフ関係にあるSOD量
癌細胞を死滅させるにはスーパーオキシドが必要なため、SOD2が過剰にありすぎると、腫瘍転移の増大につながる。
一方でSODの過剰生産は活性酸素であるスーパーオキシドを抑制し、ハエの寿命を20%増加させるなど、SODの生産量はトレードオフの関係にある。
Klothoはがん領域にとどまらない重要な遺伝子
アミロイドβへの影響 SOD2の過剰発現は、アミロイド班の沈着を減少させ、ADマウスの記憶障害を改善する。
http://www.ncbi.nlm.nih.gov/pubmed/19666610/
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2815734/
MnSODノックアウトマウスでは、脳内のアミロイドβレベルおよびアミロイドβの負荷を有意に増加させる。
http://www.ncbi.nlm.nih.gov/pubmed/15147524/
SOD2の欠損は、アミロイド負荷を悪化させ、リン酸化タウレベルを増加させる。
http://www.ncbi.nlm.nih.gov/pubmed/17579710/
アルツハイマー病の治療標的としてのSOD-2活性
http://www.ncbi.nlm.nih.gov/pubmed/16687508
パーキンソン病、レビー小体型認知症患者での高いMn-SOD発現
http://www.ncbi.nlm.nih.gov/pubmed/19298851/
MnSODの神経保護効果
http://www.ncbi.nlm.nih.gov/pubmed/9482791/
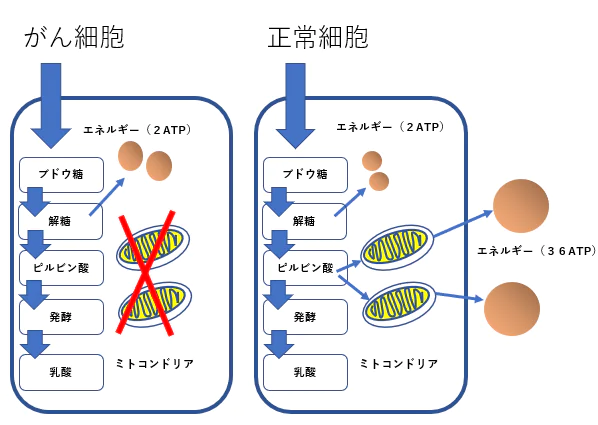
STAT3はワールブルグ効果の成立にも関与する
- がん細胞のワールブルグ効果を是正することはがん治療に有効です。その方法として2-デオキシグルコースやジクロロ酢酸やメトホルミンやケトン食などがあります。
- STAT3活性阻害方法はワールブルグ効果の是正に役立ちます。
- STAT3の活性化が低酸素誘導因子(HIF-1 )を活性化し、解糖系を亢進していることが報告されています。
STAT3 Activaties and Energy Metabolism:Dangerous Liaisons.Cancer6(3):1579- 1596 .2014
ワールブルグ効果
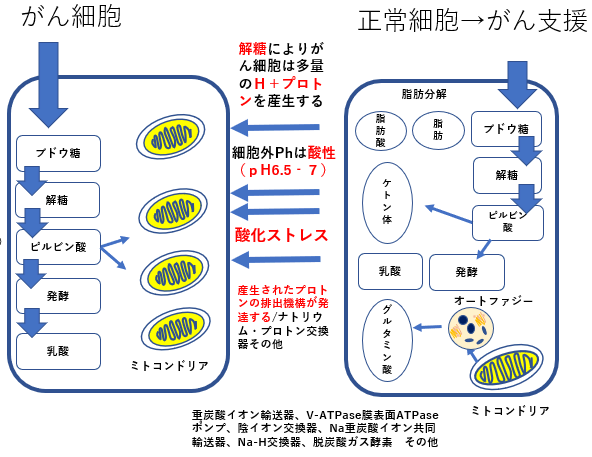
リバース・ワールブルグ効果
オーラノフィンのSTAT3阻害作用
- オーラノフィン(Auranofin)は関節リュウマチの治療に使用される経口金製剤です。通常、非ステロイド抗炎症剤を使用しても効果がないときに使われます。
- 最近、オーラノフィンの抗腫瘍効果が注目されており米国ではがん治療へのオーラノフィンの効果を検討する第2相臨床試験の実施がFDA(米国医薬品局)から承認されています。
- オーラノフィンの抗腫瘍作用のメカニズムとしては様々な作用が報告されていますが、特にチオレドキシン還元酵素の阻害による抗がん作用が注目されています。
- オーラノフィンのSTAT3の活性阻害
- オーラノフィンのSTAT3の抑制とテロメラーゼ活性の阻害
Auranofin blocks interleukin-6 signalling by inhibiting phosphorylation of JAK1 and STAT3.Immunology.122(4):607- 14,2007
ヒト肝臓がん細胞HepG32を用いた実験では、オーラノフィンはIL6によるヤーヌキナーゼ(JAK)とSTAT3のリン酸化を阻害したと報告されています。また、オーラノフィンはIL6で産生が誘導されるハプトグロビン、フィブリノーゲン、補体C3,α1-産生糖タンパクなどの急性期タンパク質(炎症の急性期に肝臓から産生されるタンパク質)の産生、および血管内皮細胞増殖因子の遺伝子発現を抑制した。これらの遺伝子の転写活性はSTAT3に依存している。
Antiproliferative effect of gold(Ⅰ)compound auranofin through inhibition of STAT3 and telomerase activity in MDA-MB231 human breast cancer cells.
BMB rep.46(1):59-64、2013
オーラノフィンが、MDA-MB231ヒト乳がん細胞のSTAT3阻害及びテロメラーゼ活性阻害に関与していことを示している。
オーラノフィンは下痢などの消化器症状やその他の副作用には十分に気をつけなければなりませんが、その作用機序と副作用を十分に理解して使用すればがん治療に利用価値が高い薬かもしれません。
文 献
1) Chapman, P.B., Hauschild, A., Robert, C., Haanen, J.B., Ascierto, P., Larkin, J., Dummer, R., Garbe, C., Testori, A., Maio, M., et al.; BRIM-3 Study Group. (2011) Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med., 364, 2507‒2516.
2) Boucher, M.J., Morisset, J., Vachon, P.H., Reed, J.C., Lainé, J., & Rivard, N. (2000) MEK/ERK signaling pathway regulates the expression of Bcl-2, Bcl-XL, and Mcl-1 and promotes survival of human pancreatic cancer cells. J. Cell. Biochem., 79, 355‒369.
3) García-Sáez, A.J. (2012) The secrets of the Bcl-2 family. Cell Death Differ., 9, 1733‒1740.
4) Lee, H., Herrmann, A., Deng, J.H., Kujawski, M., Niu, G., Li, Z., Forman, S., Jove, R., Pardoll, D.M., & Yu, H. (2009) Persistently activated Stat3 maintains constitutive NF-kappaB activity in tumors. Cancer Cell, 15, 283‒293.
5) Lee, H.J., Zhuang, G., Cao, Y., Du, P., Kim, H.J., & Settleman, J. (2014) Drug resistance via feedback activation of Stat3 in oncogene-addicted cancer cells. Cancer Cell, 26, 207‒221.
6) Su, F., Bradley, W.D., Wang, Q., Yang, H., Xu, L., Higgins, B., Kolinsky, K., Packman, K., Kim, M.J., Trunzer, K., et al. (2012) Resistance to selective BRAF inhibition can be mediated by modest upstream pathway activation. Cancer Res., 72, 969‒978.
7) Nazarian, R., Shi, H., Wang, Q., Kong, X., Koya, R.C., Lee, H., Chen, Z., Lee, M.K., Attar, N., Sazegar, H., et al. (2010) Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature, 468, 973‒937.
8) Zheng, X., Carstens, J.L., Kim, J., Scheible, M., Kaye, J., Sugimoto, H., Wu, C.C., LeBleu, V.S., & Kalluri, R. (2015) Epithelial-tomesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature, 527, 525‒530.
9) Fischer, K.R., Durrans, A., Lee, S., Sheng, J., Li, F., Wong, S.T., Choi, H., El Rayes, T., Ryu, S., Troeger, J., et al. (2015) Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature, 527, 472‒476.
10) Zheng, X., Carstens, J.L., Kim, J., Scheible, M., Kaye, J., Sugimoto, H., Wu, C.C., LeBleu, V.S., & Kalluri, R. (2015) Epithelial-tomesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature, 527, 525‒530.
11) Pastushenko, I., Brisebarre, A., Sifrim, A., Fioramonti, M., Revenco, T., Boumahdi, S., Van Keymeulen, A., Brown, D., Moers, V., Lemaire, S., et al. (2018) Identification of the tumour transition states occurring during EMT. Nature, 556, 463‒468.
12) Mani, S.A., Guo, W., Liao, M.J., Eaton, E.N., Ayyanan, A., Zhou, A.Y., Brooks, M., Reinhard, F., Zhang, C.C., Shipitsin, M., et al. (2008) The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell, 133, 704‒715
13)Lowe S W, Ruley H E, Jacks T, Housman D E. Cell. 1993;74:957–967. [PubMed] [Google Scholar]
14) Aas T, Borresen A-L, Geisler S, Smith-Sorensen B, Johnsen H, Varhaug J E, Akslen L A, Lonning P E. Nat Med. 1996;2:811–814. [PubMed] [Google Scholar]
15) Perego P, Giarola M, Righetti S C, Supino R, Caserini C, Delia D, Pierotti M A, Miyashita T, Reed J C, Zunino F. Cancer Res. 1996;56:556–562. [PubMed] [Google Scholar]
16) Fan S, El-Diery W S, Bae I, Freeman J, Jondle D, Bhatia K, Fornace A J, Jr, Magrath I, Kohn K W, O’Connor P M. Cancer Res. 1994;54:5824–5830. [PubMed] [Google Scholar]
17).Lowe S W, Bodis S, McClatchey A, Remington L, Ruley H E, Fisher D E, Housman D E, Jacks T. Science. 1994;266:807–810. [PubMed] [Google Scholar]








